Background
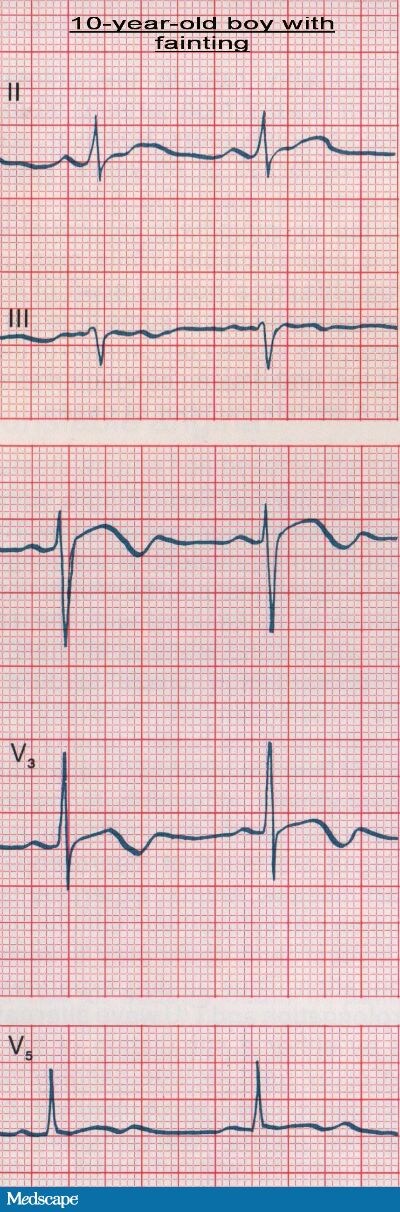 Figure 1. (Click to enlarge) |
A 10-year-old boy is brought to an outpatient pediatric clinic by his parents with a history of multiple fainting spells. These spells are sometimes complicated by generalized tonic-clonic "seizurelike" activity. The patient first started experiencing these attacks 8 months ago. His mother has noticed that the fainting most often occurs either in the early morning, after the sounding of an alarm clock, or during some type of sports activity. The child states that the seizures occur without warning, and they are sometimes associated with urinary incontinence or vomiting. According to the patient's family, the child remains unconscious for about 1 minute, after which he awakens abruptly, with no evidence of confusion and full recall of all of the events preceding the attack.
The patient was born full-term, without any complications. He has no chronic medical conditions and is not on any medications. As a result of experiencing similar symptoms, his father was diagnosed with epilepsy and started on treatment at the age of 8 years; however, the father has been without treatment and has not had any attacks since the age of 14. The patient's paternal uncle had also been diagnosed with epilepsy at the age of 10; he died during a seizure at the age of 19. The patient has an 8-year-old brother who is well, with no history of seizures or fainting spells.
On physical examination, the patient is a well-appearing and well-developed boy whose weight and height are in the 50th and 60th percentiles, respectively. His oral temperature is 98.6°F (37.0°C). His pulse is strong at 66 bpm, with a regular rhythm. His blood pressure is 105/65 mm Hg, and his respiratory rate is 15 breaths/min. The examination of his head and neck is normal. The lungs are clear to auscultation and normal respiratory effort is noted. Cardiac auscultation reveals normal S1 and S2 heart sounds, and no audible murmurs, rubs, or gallops are heard. His abdomen is soft, with no tenderness. No organomegaly is detected. The neurologic examination reveals intact cranial nerves and intact speech. Sensory and motor functions are normal in all extremities, without any pronator drift. The deep tendon reflexes are brisk and symmetric throughout. The patient's Romberg sign is negative, and his gait is stable.
The laboratory analysis, including a complete blood cell (CBC) count and a basic metabolic panel with serum electrolytes (including calcium and magnesium), is noted as normal. A chest radiograph and a computed tomography (CT) scan of the brain are also both normal. An electrocardiogram (ECG) is obtained (see Figure 1).
Discussion
| Figure 1. (Click to enlarge) | 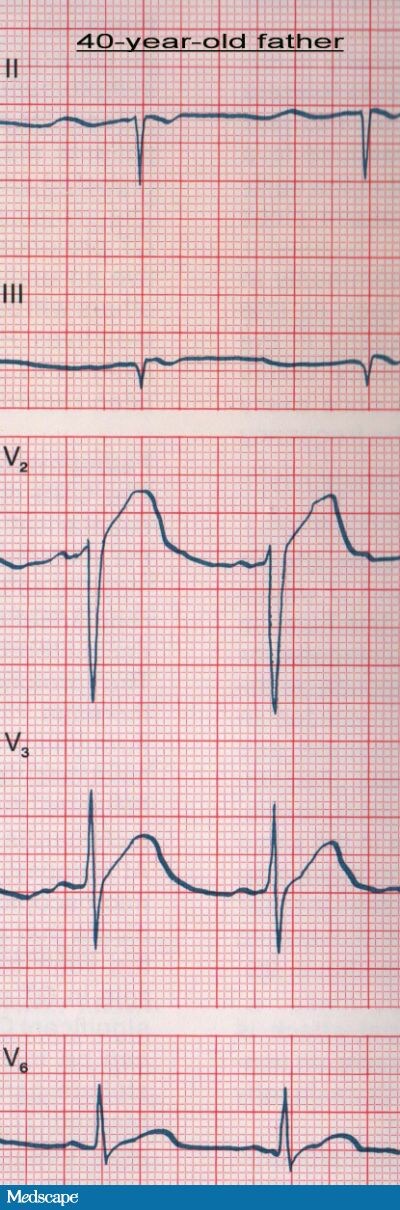 Figure 2. (Click to enlarge) |
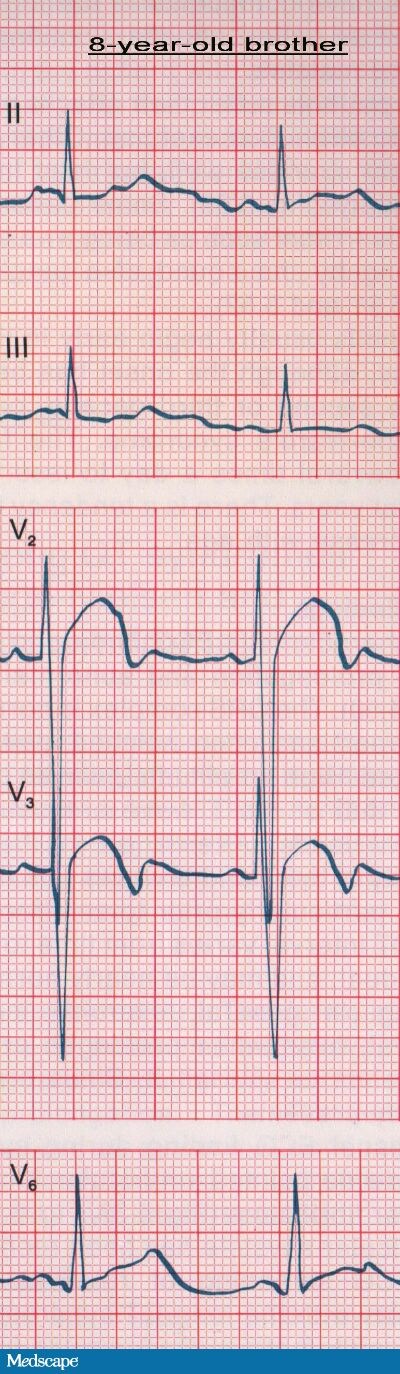 Figure 3. (Click to enlarge) | |
The patient's ECG showed a prolonged QT interval. The corrected QT (QTc) was 0.56 seconds. In addition, a biphasic T wave was seen in the precordial leads. His father's ECG (see Figure 2) also showed a prolonged QT interval (QTc=0.55 seconds) and a high-amplitude, rounded T wave in leads V2 and V3. On the ECG taken of the patient's 8-year-old brother (see Figure 3), the QTc was 0.52 seconds, and T/U wave abnormalities in leads V2 and V3 were also detected. In the context of the patient's history of recurrent fainting and the family history of "seizures" and sudden death, the prolonged QT intervals noted were very suggestive of intermittent ventricular arrhythmias caused by a congenital long QT syndrome.
Long QT syndrome (LQTS) is an electrical disease of the ventricular myocardium that is characterized by prolonged ventricular repolarization, which results in prolongation of the QT interval on the surface ECG and an increased risk of sudden death. It is characteristically associated with the potentially life-threatening cardiac arrhythmia known as "torsade de pointes", which is a form of polymorphic ventricular tachycardia.[1]
A prolonged QT interval may be acquired (usually resulting from drugs or electrolyte disturbances) or congenital. The congenital form is caused by mutations in the gene coding for the cardiac potassium, sodium, or calcium ion channels; about 400 mutations in 10 gene loci have been identified.[2] The distinct genetic types are designated LQT1-LQT10. LQT1, LQT2, and LQT3 account for over 90% of cases of LQTS, with some estimated prevalences of 45%, 45%, and 7%, respectively.[3] The specific genotype influences the clinical course, the kinds of triggering events that may initiate arrhythmias, the prognosis, and even the recommended form of treatment.[3] An underlying genetic predisposition has even been identified in some patients with the acquired form of LQTS.
Traditionally, congenital LQTS has been characterized as 2 clinical entities:
- Romano-Ward syndrome, which is inherited in an autosomal dominant fashion and only has cardiac manifestations.
- Jervell and Lange-Nielsen syndrome, which is inherited in an autosomal recessive fashion and is associated with sensorineural deafness.
Most of the epidemiologic and clinical data for congenital LQTS comes from reports from the International LQTS Registry. The registry, which began in 1979 and is still ongoing, is a major source of data about the incidence, natural history, and prognosis of patients with congenital LQTS.[14]
The incidence of congenital LQTS is difficult to determine, but it is estimated to be 1 case per 2,500-10,000 population, with most estimates in and around 1 case per 5,000 population.[4] This broad range is a result of the fact that a large number of cases go undiagnosed; about 20-50% of affected patients may not demonstrate QT prolongation on a resting ECG. Technical difficulties and methodological controversies that exist in accurately measuring the QT interval are, in part, to blame. It is, nonetheless, one of the most common causes of autopsy-negative, unexplained sudden death. Women are more commonly affected than men. Patients with congenital LQTS usually present in childhood, adolescence, or early adulthood, and they usually present with palpitations, syncope or near syncope, seizures, or cardiac arrest. Syncopal episodes associated with secondary seizures may be misdiagnosed as primary seizure disorders. The seizures are likely secondary to hypoperfusion of the brain during arrhythmic events. Cardiac dysrhythmias can be initiated by an external trigger, such as emotional stress, exercise, or sudden loud noises (ie, an alarm clock or telephone); however, this is not always the case. Ventricular arrhythmias may also occur during sleep, which is commonly seen in patients with the LQT3 genotype. In fact, the kind of triggering event is often linked to the underlying mutation, with certain triggers more commonly associated with certain genotypes. As many as 10% of patients are only diagnosed with LQTS at the time of sudden death.[5] Mortality can be as high as 70% in patients who remain untreated over a 10 year period.[3] This emphasizes the importance of presymptomatic diagnosis and treatment. Important clues indicating that a patient may have LQTS include an abnormal ECG, a family history of unexplained death, or hearing loss (which is present in around 4% of patients with LQTS).[6]
In patients with suspected congenital LQTS, the initial evaluation should be directed at calculating the QTc interval on a resting ECG. The QTc interval is the QT interval corrected for heart rate because, under normal physiologic circumstances, the actual measured QT interval adjusts with the heart rate; in other words, it is longer at slower rates and shorter at faster rates. QTc is calculated by dividing the measured QT by the square root of the R-R interval (the Bazett formula), both of which are measured in seconds.
Prolongation of the QTc interval is defined on the basis of the following age- and sex-specific criteria[1]:
| Population | QTc Reference range (in sec.) | Borderline QTc (in sec.) | Prolonged QTc (in sec.) |
|---|---|---|---|
| Males or females younger than 15 years of age | <0.44> | 0.44-0.46 s | >0.46 s |
| Males older than 15 years of age | <0.43> | 0.43-0.45 s | >0.45 s |
| Females older than 15 years of age | <0.45> | 0.45-0.46 s | >0.46 s |
For practical purposes, any value over 0.45 seconds should be considered abnormal. To increase accuracy, the QT interval can be manually measured on serial ECGs by using multiple leads, measuring several successive beats, and calculating their average for each ECG.[7] Nevertheless, approximately 10-15% of patients with congenital LQTS (diagnosed by genetic tests) present with a normal QTc duration[1]; therefore, if suspicion is high despite a normal ECG, Holter monitoring or stress testing may be necessary to try to provoke or unmask the long QTc.
Other ECG features that help to establish the diagnosis of congenital LQTS include:
- An abnormal T-wave morphology, including notched or biphasic T waves.
- The presence of T-wave alternans, which is defined as the regular alternation in T wave amplitude or polarity.
- An increased QT dispersion, which is defined as the variability in QT duration among different ECG leads.
A scoring system for the diagnosis of congenital LQTS was established in 1985 by Schwartz et al and revised in 1993, but still serves as the best guide for clinicians today.[8] It incorporates the ECG criteria (the measured resting QTc interval, history of torsades de pointes, presence of T-wave alternans or notched T wave on an ECG, and low heart rate for age), the clinical criteria (syncope or congenital deafness), and family history (family members with definite LQTS or unexplained sudden death before the age of 30). Points ranging from 0.5 to 3 are assigned to each of the above criteria, and the points are added to calculate the LQTS score. Depending on the patient's score, the probability of having LQTS is rated as low (<1>
Genetic testing for congenital LQTS is now available in specialized centers; however, the practical application of genetic testing is limited because of the complexity and heterogeneity of congenital LQTS. In addition, as many as 25% of patients have unknown mutations[9]; therefore, a negative test does not exclude the disease. Once an index case with congenital LQTS is identified, evaluation needs to extend to all first-degree relatives, and treatment must be established where indicated.
The guidelines published in 2006 by the American College of Cardiology, the American Heart Association, and the European Society of Cardiology consider "lifestyle modifications," defined as the contraindication of competitive sports and of all drugs known to prolong the QT interval, as a class I recommendation and an important strategy for the prevention of fatal arrhythmia in patients with congenital LQTS.[10] The mainstay of medical treatment for LQTS is the use of beta-blockers.[11] Beta-blockers shorten the QT interval, which decreases the risk of torsade de pointes arrhythmia and reduces the incidence of syncope and sudden cardiac death. They are effective in approximately 70% of patients. In high-risk patients, implantable cardioverter-defibrillators (ICDs) appear to be the most effective therapy. An ICD may also be considered as a primary therapy if the patient has a strong family history of sudden cardiac death.[12] Another therapeutic measure reserved for patients who continue to have symptoms despite medical therapy and ICD placement is left cervicothoracic sympathectomy (LCTS). LCTS causes sympathetic denervation of the heart and, in some cases, decreases the event rate.[12] Because of the appreciable risk of torsades de pointes arrhythmia and sudden cardiac death without treatment, all symptomatic patients with congenital LQTS should be treated. Treating asymptomatic patients is more controversial; however, because sudden cardiac death can be the first manifestation of LQTS, a safe approach would be to treat even asymptomatic patients with at least medical therapy.[1] Patients who survive a cardiac arrest, fail beta-blocker therapy, or have other high-risk features based on family history or genetic screening should be considered for an ICD. It is important to conduct any workup for LQTS in consultation with a cardiologist.
Gene-specific therapy is currently an area under investigation, and the management of LQTS is increasingly being guided by gene-specific diagnoses. General recommendations based upon knowledge of the patient's genotype that assist in making treatment selections include avoidance of strenuous exercise, stress, and unsupervised swimming or diving in patients with LQT1, because these are common triggers of arrhythmia. Beta-blockers are highly protective in these patients. LQT2 is also induced by exercise, but to a lesser degree than LQT1 is. Among patients with LQT3, events are less commonly induced by exercise; they usually occur during sleep or rest. Beta-blockers are less beneficial in these patients.[3,13] In a number of case reports, mexiletine (a sodium channel blocker) has been demonstrated to shorten the QT interval in the subgroup of patients with LQT3.
In this case, the diagnosis was confirmed by an exertional ECG recording that reproduced torsade de pointes arrhythmia. The patient was started on a beta-blocker and has since remained asymptomatic. Because of the high risk-profile in this family, his younger brother was also started on beta-blocker therapy.
The mainstay of therapy in symptomatic and asymptomatic patients with congenital LQTS is beta-blockers. Beta-blockers shorten the QT interval, which decreases the risk of torsade de pointes arrhythmia and reduces the incidence of syncope and sudden cardiac death. They are effective in approximately 70% of patients; however, 30% of patients continue to experience cardiac events. In this group of high-risk patients, implanted cardioverter-defibrillators (ICDs) appear to be the most effective therapy. Mexiletine, a sodium channel blocker, may help in shortening the QT interval in the LQT3 subgroup of patients and improve protection.e. Class 1 antyarritmic agent

No comments:
Post a Comment